|
Hereditary Ataxias are an inherited, or genetic, disorder. That means that it is caused by an abnormality of a single gene. To understand how the disease is passed on, it is important to know about genes and cells. Each gene is like a blueprint that tells the cell how to make a certain chemical or protein (like heart muscle proteins, or neurotransmitter, or eye color pigments). It is estimated that humans have about 100,000 genes inside each of their body cells; not all genes are active in all the cells (for instance, a gene for heart muscle protein doesn't need to be active in a brain cell). Each egg cell that a woman makes contains one copy of each of the 100,000 genes, and one copy of each of the father's genes is contained in his sperm cell, so that when a new person arises from the joining of an egg and a sperm cell, the new person has two copies of each gene (one from his father, and one from his mother). This genetic pattern is called "autosomal recessive inheritance", which means that 1) the disease is hereditary, 2) a double dose of the altered or non functioning gene is required to cause symptoms, 3) the disease can strike males and females with equal likelihood, and 4) that it is possible to "carry" the altered gene without having symptoms of the disease. All people have genes that do not work properly (think of them as blueprints with a window or a door missing). In the case of Friedreich's ataxia, and a number of other hereditary diseases, symptoms do not appear unless a person has a double dose of an altered, or non functioning, Friedreich's gene. People who have one copy of an altered "Friedreich's gene" are called "carriers" of Friedreich's ataxia and can pass their altered gene on to their children, but these carriers do not have any symptoms of Friedreich's ataxia. That is because the second copy of the Friedreich's gene is still working, and that is enough to prevent any symptoms from occurring. It is only people who get a "double dose" of the altered gene, so that neither copy of their "Friedreich's genes" works properly, who develop symptoms that are recognized as Friedreich's ataxia. In the United States, it has been estimated that 1 out of every 100 people is a carrier of the (altered) Friedreich's gene. In some regions or ethnic groups, this number may be a little higher or lower, but if you look around you at a busy airport or football game, there are probably several people there who are carriers AND DON'T KNOW IT. Most of the time, the development of Friedreich's in a child comes as a complete surprise to their parents, who had no way of knowing that they were carriers of the Friedreich's gene.
The most common flaw (mutation) in the frataxin gene that leads to FRDA is known as a "triplet repeat." In this type of mutation, a section of DNA is repeated over and over again, from as many as 100 to more than 1,000 times. The presence of this extra bit of genetic material interferes with the normal production of frataxin protein. Research has shown that the number of repeat units varies among people with FRDA, and even among tissues in the same person. Recent research indicates that people with smaller numbers of repeats generally have later disease onset, slower progression and less severe heart problems. However, there is too much variability in the course of the disease among people with similar numbers of repeats to allow predictions about disease severity from this factor alone.
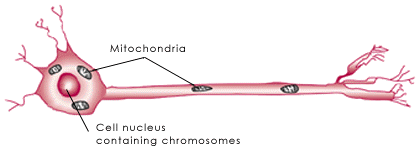
Recent research has revealed that the probable role of the frataxin protein in cells is to regulate the amount of iron located in the cells' mitochondria, tiny energy-producing units found inside the cells (see illustration, below).
The most common flaw (mutation) in the frataxin gene that leads to FRDA is known as a "triplet repeat." In this type of mutation, a section of DNA is repeated over and over again, from as many as 100 to more than 1,000 times. The presence of this extra bit of genetic material interferes with the normal production of frataxin protein. Research has shown that the number of repeat units varies among people with FRDA, and even among tissues in the same person.
A number of things are very important for a person with Friedreich's and his or her family to do. Most importantly, they should have a thorough evaluation by a physician or neurologist who is sensitive to all the possible complications of Friedreich's. An individual with Friedreich's may need a neurologic examination and tests (which may include evaluation of the cerebellum and spine cord with CT (computerized tomography) or MRI (magnetic resonance imaging), studies of the peripheral nerves with EMG (electromyography), and other studies to rule out other conditions; an ophthalmologic (eye) exam, an audiologic (hearing) test, evaluation and treatment by an orthopedist (bone doctor), an EKG (electrocardiogram), and perhaps evaluation by an endocrinologist (diabetes, thyroid doctor). Evaluation or treatment by a speech pathologist, physical or occupational therapist, and urologist may become necessary as the disease progresses. Patients and families with Friedreich's ataxia should undergo genetic counseling. Family members usually have many questions about the chances that other children might get or have the disease, or be carriers of the disease gene. These questions, as well as questions about the rapidly evolving technology for genetic testing for Friedreich's can be answered by a genetic counselor. |
|
Click the 'Back' button on your browser's toolbar to go BACK TO MY MAIN PAGE |